- Phare
- Validé par KD/KO
Anticorps Polyclonal de lapin anti-SMN
SMN Polyclonal Antibody for WB, IHC, IF/ICC, IP, ELISA
Hôte / Isotype
Lapin / IgG
Réactivité testée
Humain, rat, souris
Applications
WB, IHC, IF/ICC, IP, ELISA
Conjugaison
Non conjugué
N° de cat : 11708-1-AP
Synonymes
Galerie de données de validation
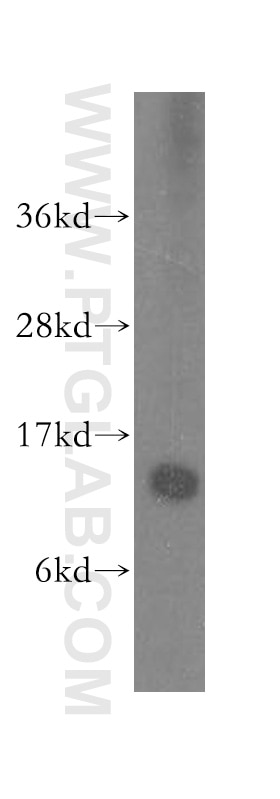
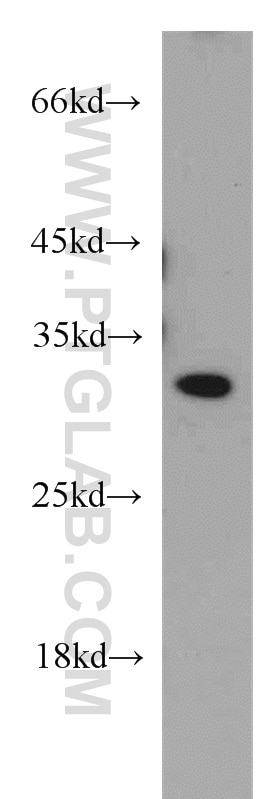
 with sh-Control and sh-SMN transfected HeLa cells.")
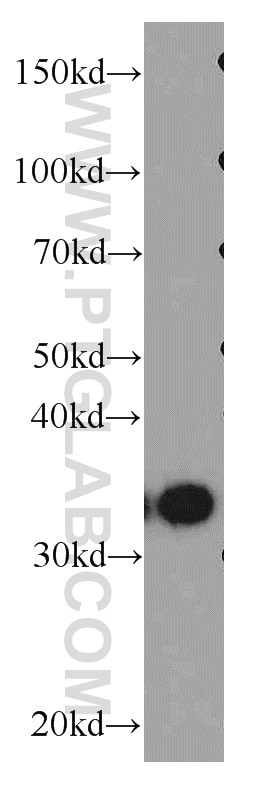
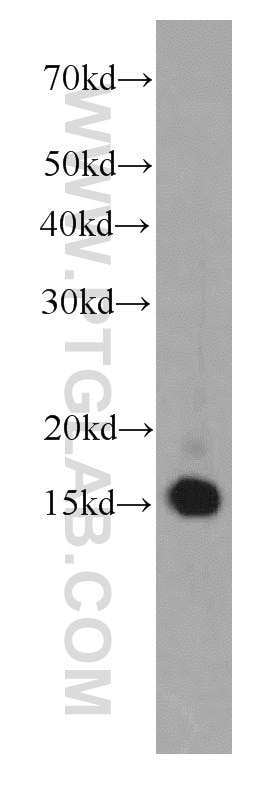
 at dilution of 1:8000 incubated at room temperature for 1.5 hours.")
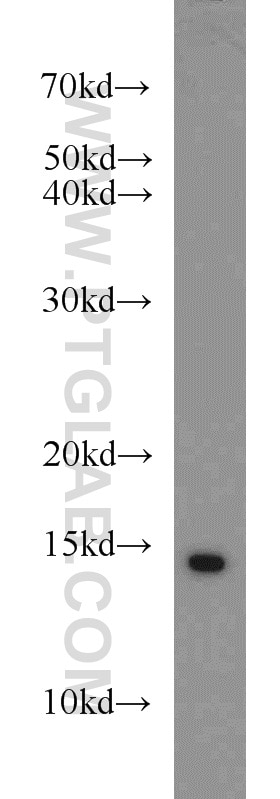
 at dilution of 1:1000 incubated at room temperature for 1.5 hours.")
 at dilution of 1:1500 incubated at room temperature for 1.5 hours.")
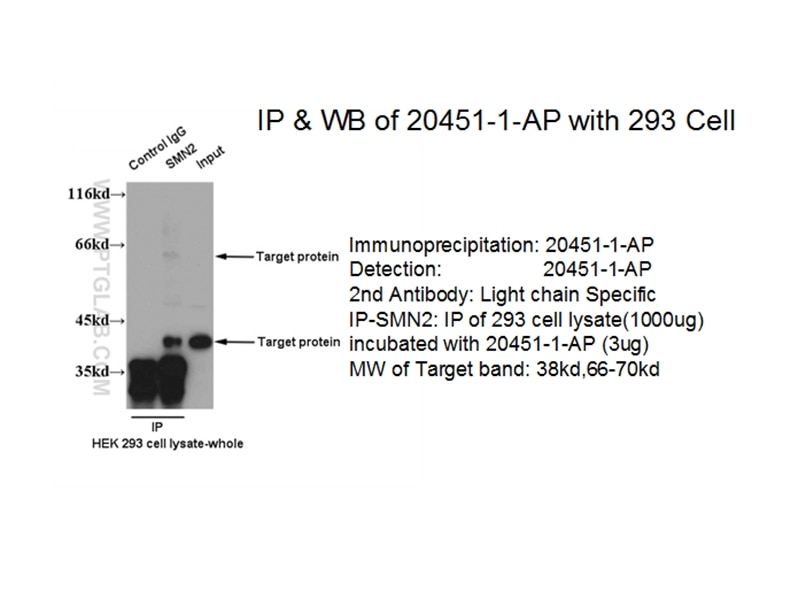
 with HEK-293 cells lysate 1040 ug.")
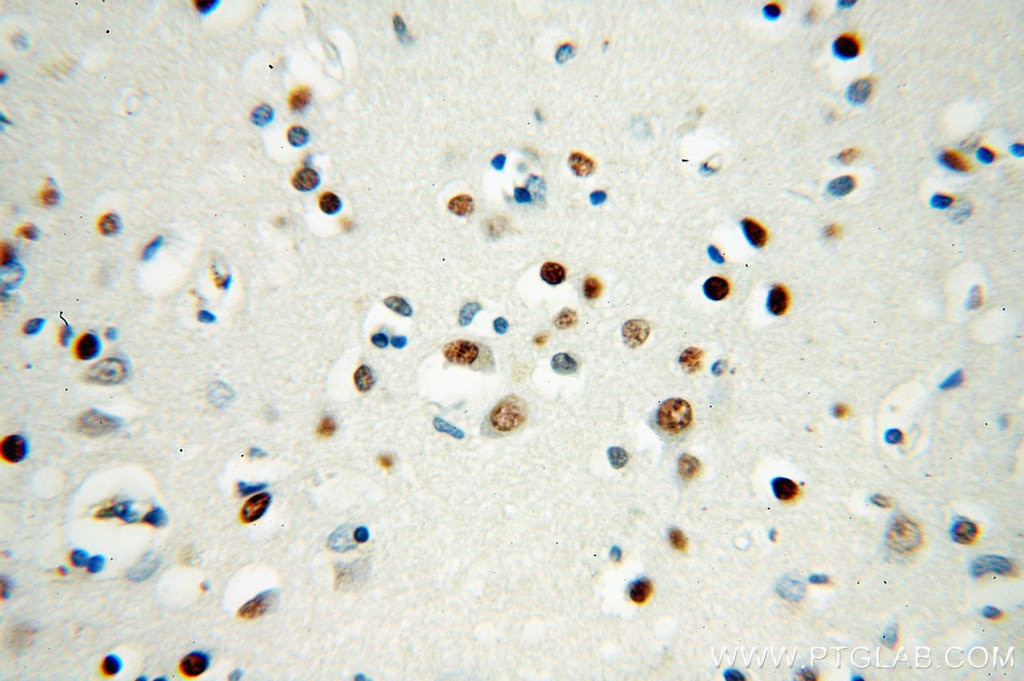
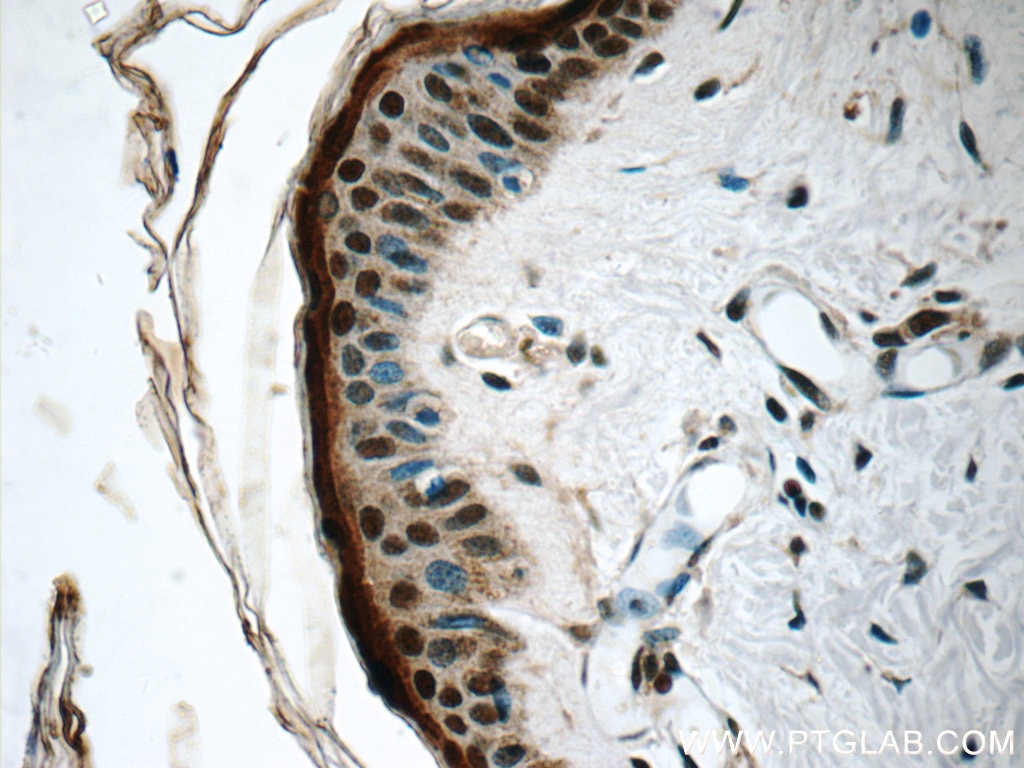
 at dilution of 1:100 (under 10x lens).")
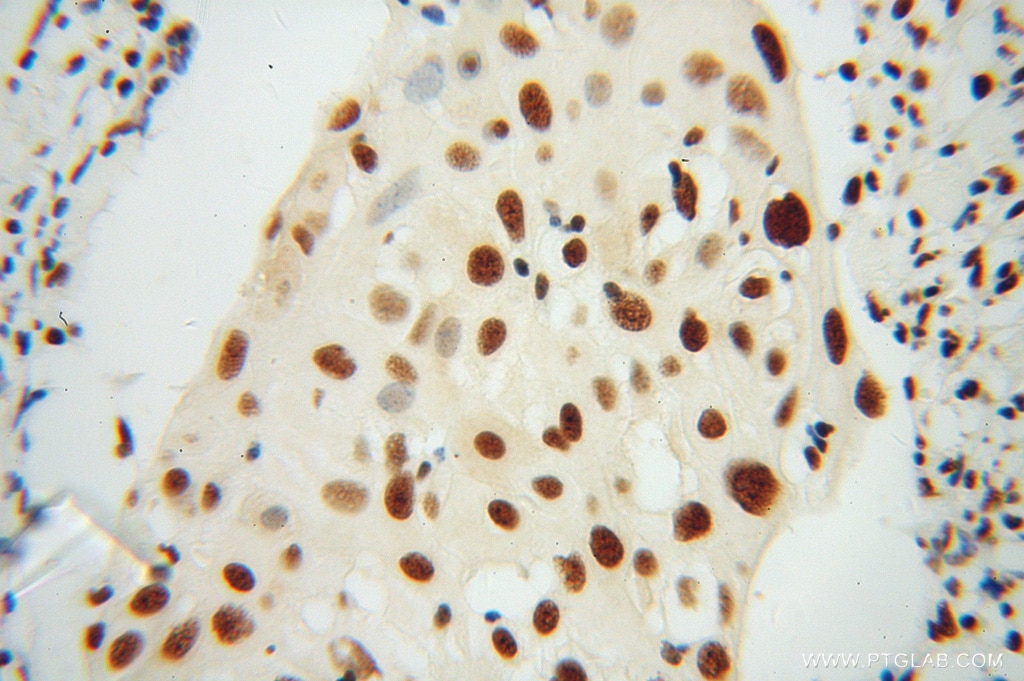
 at dilution of 1:100 (under 40x lens).")
 at dilution of 1:100 (under 40x lens).")
 at dilution of 1:100 (under 40x lens).")
 at dilution of 1:100 (under 40x lens).")
 at dilution of 1:100 (under 40x lens).")
 at dilution of 1:100 (under 40x lens).")
 at dilution of 1:100 (under 40x lens).")
 at dilution of 1:100 (under 40x lens).")
 at dilution of 1:50 (under 40x lens).")
 fixed HepG2 cells using SMN antibody (11708-1-AP) at dilution of 1:800 and CoraLite®488-Conjugated AffiniPure Goat Anti-Rabbit IgG(H+L), CL594-Phalloidin (red).")
Applications testées
| Résultats positifs en WB | cellules HEK-293, cellules HeLa, cellules HepG2, cellules Jurkat, cellules K-562, tissu testiculaire de souris |
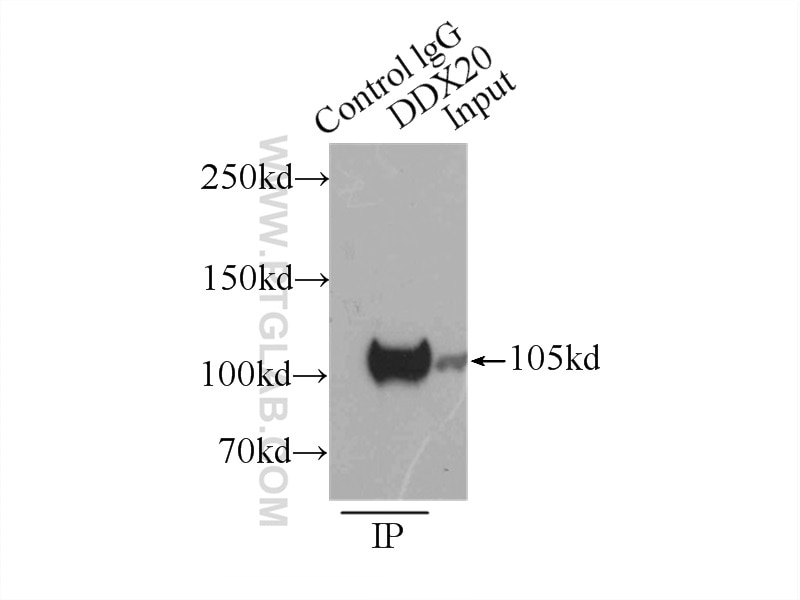
| Résultats positifs en IP | cellules HEK-293, |
| Résultats positifs en IHC | tissu rénal humain, tissu cardiaque humain, tissu cérébral humain, tissu cutané humain, tissu ovarien humain, tissu placentaire humain, tissu pulmonaire humain, tissu splénique humain, tissu testiculaire humain il est suggéré de démasquer l'antigène avec un tampon de TE buffer pH 9.0; (*) À défaut, 'le démasquage de l'antigène peut être 'effectué avec un tampon citrate pH 6,0. |
| Résultats positifs en IF/ICC | cellules HepG2, |
Dilution recommandée
| Application | Dilution |
|---|---|
| Western Blot (WB) | WB : 1:2000-1:16000 |
| Immunoprécipitation (IP) | IP : 0.5-4.0 ug for 1.0-3.0 mg of total protein lysate |
| Immunohistochimie (IHC) | IHC : 1:50-1:200 |
| Immunofluorescence (IF)/ICC | IF/ICC : 1:400-1:1600 |
| It is recommended that this reagent should be titrated in each testing system to obtain optimal results. | |
| Sample-dependent, check data in validation data gallery | |
Applications publiées
| KD/KO | See 1 publications below |
| WB | See 3 publications below |
| IF | See 3 publications below |
| IP | See 1 publications below |
| ELISA | See 4 publications below |
Informations sur le produit
11708-1-AP cible SMN dans les applications de WB, IHC, IF/ICC, IP, ELISA et montre une réactivité avec des échantillons Humain, rat, souris
| Réactivité | Humain, rat, souris |
| Réactivité citée | rat, Humain, souris |
| Hôte / Isotype | Lapin / IgG |
| Clonalité | Polyclonal |
| Type | Anticorps |
| Immunogène | SMN Protéine recombinante Ag2260 |
| Nom complet | survival of motor neuron 2, centromeric |
| Masse moléculaire calculée | 282 aa, 30 kDa |
| Poids moléculaire observé | 38 kDa |
| Numéro d’acquisition GenBank | BC000908 |
| Symbole du gène | SMN |
| Identification du gène (NCBI) | 6607 |
| Conjugaison | Non conjugué |
| Forme | Liquide |
| Méthode de purification | Purification par affinité contre l'antigène |
| Tampon de stockage | PBS avec azoture de sodium à 0,02 % et glycérol à 50 % pH 7,3 |
| Conditions de stockage | Stocker à -20°C. Stable pendant un an après l'expédition. L'aliquotage n'est pas nécessaire pour le stockage à -20oC Les 20ul contiennent 0,1% de BSA. |
Informations générales
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disease characterized by loss of anterior horn cells in the spinal cord and concomitant symmetrical muscle weakness and atrophy (PMID: 16364894 ). SMA is caused by deletion or mutations of the survival motor neuron (SMN1) gene. SMA patients lack a functional SMN1 gene, but they possess an intact SMN2 gene, which though nearly identical to SMN1, is only partially functional (PMID: 17355180). A large majority of SMN2 transcripts lack exon 7, resulting in production of a truncated, less stable SMN protein (PMID: 10369862). The level of SMN protein correlates with phenotypic severity of SMA. This antibody, 11708-1-AP, raised against the recombinant full-length human SMN2 protein, recognizes all isoforms of SMN protein.
Protocole
| Product Specific Protocols | |
|---|---|
| WB protocol for SMN antibody 11708-1-AP | Download protocol |
| IHC protocol for SMN antibody 11708-1-AP | Download protocol |
| IF protocol for SMN antibody 11708-1-AP | Download protocol |
| IP protocol for SMN antibody 11708-1-AP | Download protocol |
| Standard Protocols | |
|---|---|
| Click here to view our Standard Protocols |
Publications
| Species | Application | Title |
|---|---|---|
Nat Commun SNUPN deficiency causes a recessive muscular dystrophy due to RNA mis-splicing and ECM dysregulation | ||
Hum Mol Genet SMN expression is required in motor neurons to rescue electrophysiological deficits in the SMNΔ7 mouse model of SMA. | ||
Hum Mol Genet Low levels of Survival Motor Neuron protein are sufficient for normal muscle function in the SMNΔ7 mouse model of SMA. | ||
Neurobiol Dis Dual SMN inducing therapies can rescue survival and motor unit function in symptomatic ∆7SMA mice. | ||
Hum Mol Genet Intragenic complementation of amino and carboxy terminal SMN missense mutations can rescue Smn null mice.
| ||
Hum Mol Genet Mild SMN missense alleles are only functional in the presence of SMN2 in mammals. |
Avis
The reviews below have been submitted by verified Proteintech customers who received an incentive forproviding their feedback.
FH Rachel (Verified Customer) (08-19-2019) | Worked well in MSD immunoassays and cell based assays.
|