Guide to Flow Cytometry Panel Building
Written by Dr. Frédéric Duval and Sari Gezzar-Dandashi from the Maisonneuve-Rosemont Hospital Research Center.
Flow cytometry is capable of dissecting cellular populations in detail by analyzing thousands of cells per second. For that reason, a growing number of scientific fields is now employing this indispensable application. Your flow panel is key to measuring the expression levels of these cells accurately. The challenge lies in constructing a precise and effective flow cytometry panel. Here, we will guide you through this intricate process with insights that can benefit both novice and seasoned researchers.
1. Clearly Define Markers for Your Biological Question
Lineage Markers: These are pivotal for identifying your populations of interest. For example, we use CD3 to define the T cell population, while CD8 and CD4 help us differentiate between CD4 (CD3+CD4+CD8-) and CD8 (CD3+CD8+CD4-) T cell populations.
Exclusion Markers: These are used to exclude cells you’re not interested in to improve signal resolution. For instance, when analyzing NK cells in PBMCs, consider adding T cell and B cell markers to remove those cell types from the analysis. (Figure 1)
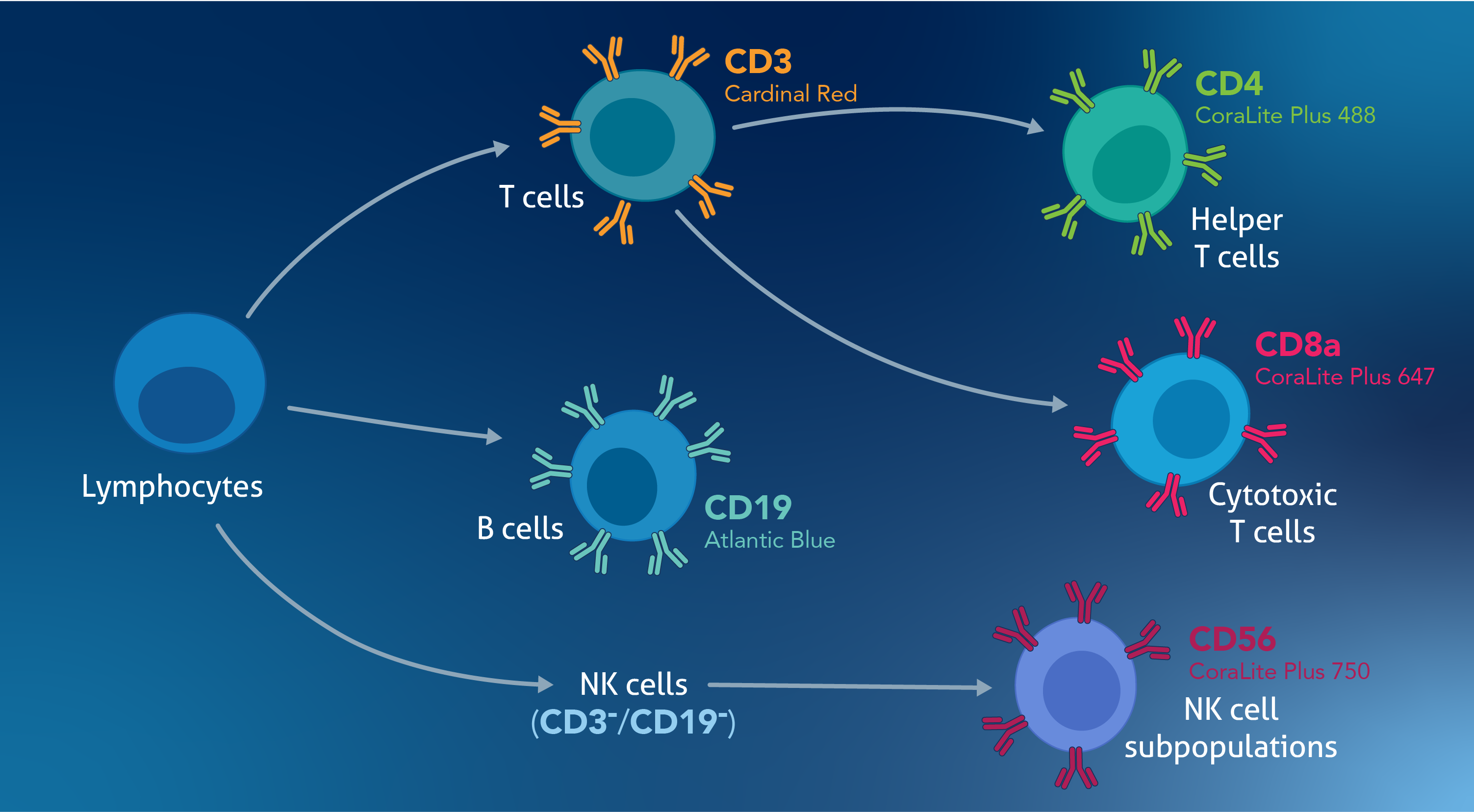
Figure 1. Adding CD3 and CD19 as exclusion markers, as in this TBNK Basics Panel (PK30012), can help exclude B cells and T cells from analysis if, for instance, your target cell population is NK cells.
Markers of Interest: These directly answer your research question and include activation markers, differentiation markers, DNA content dyes, etc.
|
Tip 1: Don’t start from scratch whenever possible There are many resources, such as Optimized Multicolor Immunophenotyping Panel (OMIP) articles, where you can consult published protocols for your populations of interest and learn of different antibody clones used by other researchers. These can also help you gather information about panel design, reagent information, titrations, and gating strategies. |
2. Know Your Instrument
-
It is essential to understand the cytometer’s detector count, optical filter configuration, the number of lasers, and their respective excitation wavelengths. The excitation spectra of your fluorochromes must align with an available laser in the system. Furthermore, the peak of the fluorochrome’s emission spectrum should coincide with an optical filter set in the cytometer to enable accurate capture and interpretation of the fluorescence signal. Each machine could have a different configuration, so ask your flow cytometry core or consult the instrument document. As a last resort, you can always contact the company with the serial number to request the information for your specific machine.
-
Use cytometers with more lasers to minimize spectral overlap. This is especially important when dealing with larger panels to decrease spillover and enhance resolution.
-
Make sure the performance and calibration of the cytometer are optimal.
3. Antibodies and Fluorochrome Selection
-
Choose clones cited in published research for outstanding performance. If this is not feasible, test multiple clones and select the one that best distinguishes the negative and positive populations (i.e., staining index). Additionally, use reference populations, cell lines validated in the literature, and/or isotype controls to assess the specificity of your clone for its target. For example, you can use a knock-out cell line as a negative control.
-
Classify the markers in your panel based on their expression level, then assign brighter fluorochromes to lowly expressed markers or those of unknown expression levels. In contrast, dimmer fluorochromes should be paired with more highly expressed markers. This will allow for the best resolution for the markers of interest while still being able to define the various populations in the sample.
-
Choose fluorochromes that can be excited by different lasers and exhibit minimal emission spectrum spillover, particularly for co-expressed markers. This approach will mitigate the spillover spreading error (Trumpet Effect, see Tip #2), a key factor contributing to reduced resolution (see Figure 2). Consult your core facility manager for a cytometer-specific spillover spreading matrix to predict and avoid fluorochrome combinations that create the Trumpet Effect.
-
Avoid pairing tandem dyes with hierarchical markers like CD8, CD3, CD4, etc., especially when a marker of interest is linked with the donor fluorochrome of the tandem. For instance, it's best to avoid coupling APC-Cy7 with a CD8 marker if one of the markers of interest is already linked to APC. Tandem dyes inherently involve a fraction of absorbed energy that may not transfer efficiently from the donor (APC) to the acceptor (Cy7). Consequently, APC from the tandem dye will emit light, potentially causing signal spreading and the generation of false positive signals in the marker of interest5.
|
Tip 2: Minimize the Spillover Spreading Error (Trumpet Effect) To address and minimize spreading during panel design or troubleshooting, three key observations can guide us:
Fluorochromes emit light across a broad spectrum, often leading to overlap in their emission spectra when multiple fluorochromes are present in a panel1. This overlap can result in the light emitted by one fluorochrome spilling into a detector intended for another. Detectors collecting spilled light from a fluorochrome are termed secondary detectors, whereas primary detectors capture photons emitted by the intended fluorochrome. The amount of spilled signals into a detector can be measured and subtracted from the total number of photons detected to only consider the light emitted by the expected fluorochrome using a mathematical function called compensation2-5. However, spilled emission into the secondary detector can cause loss in sensitivity. Spilled signals introduce inaccuracy in photon counting by the secondary detector7. Generally, spillover spreading error tends to be higher for fluorochromes with lower photon emission efficiency, i.e., the efficiency with which absorbed photons are converted to emitted photons. This tendency is often intensified when fluorochromes emit photons in wavelength ranges that are not optimally detected, such as infrared wavelengths in photon multiplier tubes8. Consequently, using compensation values for spillover spreading error prediction is, at best, of mediocre reliability. The loss of resolution can significantly impede the distinction between positive and negative signals, particularly affecting markers with a continuous and low expression pattern, often observed for markers of interest9-12. |
-
Whenever possible, run an unstained sample on the selected cytometer before assigning fluorochromes to identify the autofluorescence signature of your samples. Avoid channels where autofluorescence signals are too high. If this is impossible, assign a bright fluorochrome in this channel, as decreased resolution is expected.
-
Use a “dump channel” for exclusion markers. This strategy involves selecting a single fluorochrome for all the markers intended for exclusion. By doing so, all undesired cells that express any of these markers can be simultaneously identified and then excluded from the analysis.
-
To improve data quality, incorporate a viability dye to exclude dead or dying cells that generate aberrant signals.
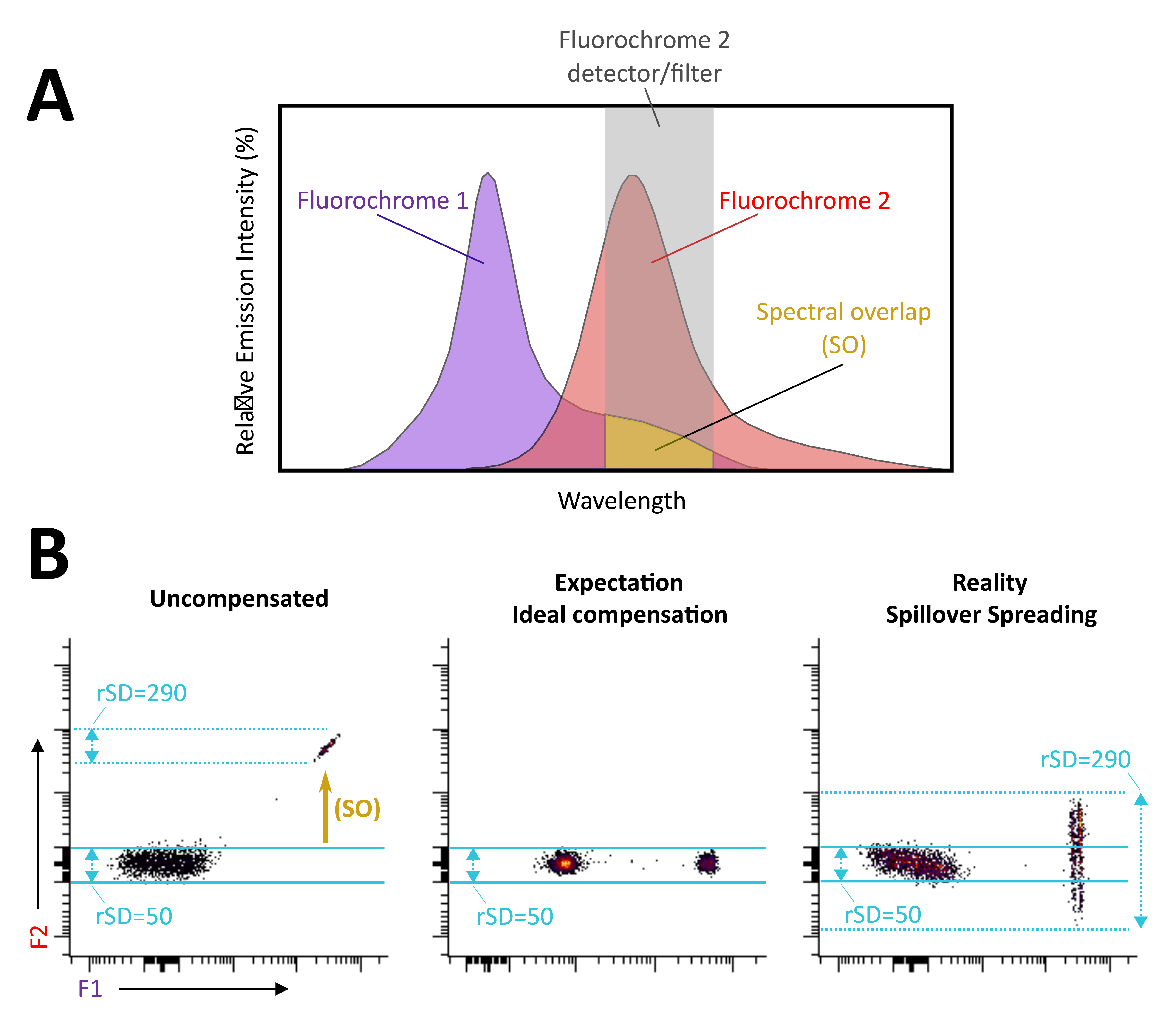
Figure 2. Spillover Spreading Error (Trumpet Effect) may occur even after proper compensation. A) The emission spectrum of fluorochromes 1 and 2 (F1 and F2) is depicted, along with a representation of the optical filter placed before the F2 detector. Due to spectral overlap, the F2 detector captures signals coming from the F1 fluorochrome. (B, left panel) Before compensation, the spillover signal from F1 is observed in the F2 detector when beads incubated solely with F1 are introduced into the cytometer. Robust standard deviation (rSD), also called spread, in the F2 detector, is 50 for the F1 negative population and 290 for the F1 positive population. (B, middle panel) Post-compensation, the contribution of F1 fluorochrome into the F2 detector has been appropriately subtracted. When spread does not occur, we expect to obtain a compensated F1 positive population exhibiting the same value in rSD compared to the negative; here, it would be 50. (B, right panel) When spillover spreading error (the Trumpet Effect) occurs, even after proper compensation, the F1 positive signal is “spread” into the F2 detector beyond the blue lines. Consequently, markers with a continuous and/or low expression pattern stained with F2 may pose challenges in detection. In such cases, a fluorescence minus one (FMO) sample, wherein only the F2 antibody is absent, can aid in distinguishing between negative and positive populations. Replacing F1 with another fluorochrome that will cause less spread is recommended if this is insufficient for precise definition.
4. Seek Assistance for Panel Design
Software Tools:
-
Panel design tools, such as EasyPanel and Fluorofinder, allow the plotting of different fluorochromes’ excitation and emission spectra and visualization of any possible spectral overlap. They also provide information about fluorochrome brightness. These tools now also have artificial intelligence that can propose fluorochrome combinations based on commercially available antibodies, the specific configuration of your cytometer, and the expected antigen density of your targets of interest.
Online Resources:
-
Many companies offer comprehensive guides with detailed instructions for panel building. YouTube channels, including but not limited to AJA Rieger Flow, Expert Cytometry, OpenFlow Cytometry, and the University of Chicago, can provide a solid basis on how cytometers work and how to use them. Such content goes beyond text guides, offering easy-to-consume videos that enhance learning, making it ideal for beginners. The University of Chicago website also provides a broad range of information, from experimental design, sample preparation, and data analysis to blogs, podcasts, and instrument demos.
-
Information on marker expression and co-expression levels can be found in the Human Protein Atlas, Antibodypedia, antibody vendor websites, and published articles.
5. Review, Test, and Refine Your Panel
-
Titrate all antibodies/fluorescent dyes to choose the dilution that maximizes the staining index without too much background noise (non-specific binding) while avoiding wasting antibodies.
-
Determine the optimal single stain controls for your samples. Choose between cells or beads. Aim for a single stain signal that matches or is brighter than the sample for proper compensation.
-
For efficiency, fully research and design your panel from the get-go instead of breaking it down into smaller segments. Adding antibodies/fluorochromes to a panel may cause unwanted consequences. Thus, optimizing the full panel from the beginning will avoid wasting time and resources.
-
Conduct a panel test with the correct controls. Check to ensure populations are identified correctly and that compensation controls are adequately correcting for any spillover and to make sure no spillover spreading is affecting the data quality. It may be necessary to change antibodies/fluorochromes to resolve these issues.
Constructing an effective flow cytometry panel demands a deep understanding of your instrument and a well-thought-out selection of markers and fluorochromes. We have walked through the critical steps of the panel design process, highlighting the importance of resource utilization and strategic planning. The challenges of spectral overlap and spillover, while inherent to the application, can be mitigated through careful design and constant iteration of your panel. The wealth of tools and resources readily available online can equip even a beginner with tools to navigate the complexities of flow cytometry.

References:
- Sahir F, Mateo JM, Steinhoff M, Siveen KS. Development of a 43 color panel for the characterization of conventional and unconventional T-cell subsets, B cells, NK cells, monocytes, dendritic cells, and innate lymphoid cells using spectral flow cytometry. Cytometry A. 2020 Dec 18.
- Roederer, M. Compensation in flow cytometry. Curr. Protoc. Cytom. 22, 1.14.1-1.14 (2002).
- Roederer, M. Spectral compensation for flow cytometry: Visualization artifacts, limitations, and caveats. Cytometry 45(3), 194–205 (2001).
- Maciorowski, Z., Chattopadhyay, P. K. & Jain, P. Basic multicolor flow cytometry. Curr. Protoc. Immunol. 117, 5.4.1-5.4.38 (2017).
- Maecker, H. T., Frey, T., Nomura, L. E. & Trotter, J. Selecting fluorochrome conjugates for maximum sensitivity. Cytometry A 62(2), 169–173 (2004).
- Ashhurst, T. M., Smith, A. L. & King, N. J. C. High-dimensional fluorescence cytometry. Curr. Protoc. Immunol. 119, 81–838 (2017).
- Nguyen, R., Perfetto, S., Mahnke, Y. D., Chattopadhyay, P., & Roederer, M. (2013). Quantifying spillover spreading for comparing instrument performance and aiding in multicolor panel design. Cytometry. Part A : The Journal of the International Society for Analytical Cytology, 83(3), 306–315.
- Bhowmick, D., van Diepen, F., Pfauth, A. et al. A gain and dynamic range independent index to quantify spillover spread to aid panel design in flow cytometry. Sci Rep 11, 20553 (2021).
- Liechti, T., Weber, L.M., Ashhurst, T.M. et al. An updated guide for the perplexed: cytometry in the high-dimensional era. Nat Immunol 22, 1190–1197 (2021).
- Mair F, Tyznik AJ. High-Dimensional Immunophenotyping with Fluorescence-Based Cytometry: A Practical Guidebook. Methods Mol Biol. 2019;2032:1-29.
- Ferrer-Font L, Pellefigues C, Mayer JU, Small SJ, Jaimes MC, Price KM. Panel Design and Optimization for High-Dimensional Immunophenotyping Assays Using Spectral Flow Cytometry. Curr Protoc Cytom. 2020 Mar;92(1):e70.
- Ashhurst TM, Smith AL, King NJC. High-Dimensional Fluorescence Cytometry. Curr Protoc Immunol. 2017 Nov 1;119:5.8.1-5.8.38.
About the authors:
 |
|
|
Dr. Frédéric Duval is a Flow Cytometry Core Manager at the Maisonneuve-Rosemont Hospital Research Center. Previously, Dr. Duval completed his Ph.D. at the University of Montreal, where he studied the role of the Notch signaling pathway in the CD8 T cell response following acute and chronic infections. He then joined the development team at CellCarta as a Data Analyst, where he evaluated the quality of designed panels, enacted gating strategies, and created analysis templates used for clinical studies. Now, as Core Manager, Frédéric has been implementing new technologies in the platform and is using his expertise to analyze and sort various cell types in different fields, such as immunology and cancerology, among others. In collaboration with his colleague Anne-Marie Aubin, he put in place novel training initiatives and educational programs and ensures that the ever-evolving best practices in flow cytometry are implemented. |
Sari Gezzar-Dandashi is a 5th year PhD candidate in Molecular Biology at the University of Montreal and is conducting his research in the laboratory of Dr. Hugo Wurtele and Dr. Elliot Drobetsky at the Maisonneuve-Rosemont Hospital Research Center. He uses flow cytometry to detect and quantify subpopulations in ovarian cancer cell lines that respond differently to DNA-damaging agents based on their expression of cell-surface markers. |


Related Content
Introduction to t-SNE for Flow Cytometry
Ask a Flow Cytometry expert: your questions answers
Support
Newsletter Signup
Stay up-to-date with our latest news and events. New to Proteintech? Get 10% off your first order when you sign up.