
Immunofluorescence techniques in zebrafish retina
Written by Natalia Jaroszynska, PhD Candidate at UCL Institute of Ophthalmology
The zebrafish (Danio rerio) is an excellent model organism for the study of development, thanks to its rapid ex vivo development, optical transparency, and many other experimental advantages. This is particularly true for tissues such as the eye, which are easily accessible for microscopy given their anatomical location. The structure and cellular composition of the zebrafish retina is almost identical to that of humans, enabling us to detect plenty of well-characterized and conserved markers with antibodies, and label specific cells or structures of interest to better understand the mechanisms of their development. Together, these make the zebrafish a fantastic model to study conserved developmental mechanisms behind retinogenesis.
Immunostaining is one of the most widely used research techniques worldwide. While plenty of reliable standard protocols have been published, our lab, over the years, has optimized and perfected this technique for staining zebrafish retina. In this blog, we provide a comprehensive review of immunofluorescence (IF) steps in the zebrafish retina, including useful tips and tricks that can be applied to any tissue of interest.
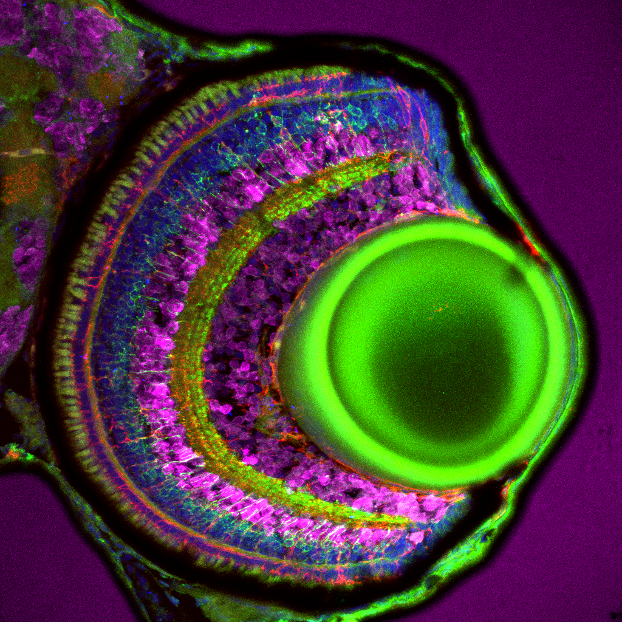
Figure 1: Immunofluorescence analysis of bipolar cells labelled with pan-PKC polyclonal antibody (12919-1-AP, green) on tissue sections of zebrafish retina at 5 days post fertilization. Dilution 1:300. Tissue fixed overnight in 4% PFA. Müller glia, red; DAPI, blue; HuC/D, magenta. Data generated by Natalia Jaroszynska in Dr. Ryan MacDonald’s lab, University College London, UK.
Basic principles of immunostaining
Immunostaining is a protein detection method that utilizes antigen-specific antibodies, which can be applied directly to tissues, allowing for the visualization of cells or subcellular structures of interest in situ. The basic premise of indirect IF is that first, a sample is incubated with a mixture of primary antibodies of interest, each raised against a specific antigen of interest, in a different host species. Next, tissues are incubated with secondary antibodies that detect primary antibodies raised in a particular host species. The secondary antibodies have a covalently conjugated fluorophore tag of a specific wavelength, allowing for their visualization of the immunolabelled proteins of interest using standard fluorescence microscopy (Figure 2).
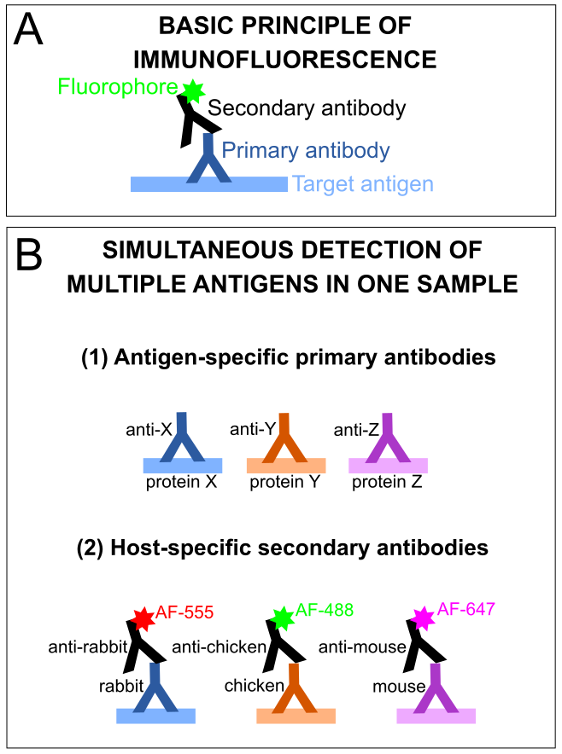
Figure 2: Basic principles of immunofluorescence and antibody-antigen detection
Before you start
Tissue preparation
Arguably the most important part of any experiment is the planning and preparation ahead of time. For the best staining results, the quality of your tissue is of utmost importance, so always use the freshest tissue you can, or ensure you store samples appropriately for your downstream applications. Secondly, the fixation of your tissue is key, so ensure you use freshly prepared, or freshly thawed (no freeze-thawing!) 4% paraformaldehyde (PFA) to achieve the best results. When incubating your zebrafish tissue or larvae in the fixative overnight at 4°C, do so on a gentle shaker to ensure that fixation is homogenous (reacted evenly across the tissue). After this stage, what you do next depends on the downstream applications. For immunostaining on frozen tissue sections, the fixed samples must be incubated in 30% sucrose solution, which acts as a cryoprotectant. We recommend conducting this step at least overnight in the fridge, before embedding in a cryomatrix and heading to the cryostat.
Expert tip: Do not store your tissue in sucrose for longer than a week to prevent bacterial or fungal growth and degradation of your proteins of interest. For long-term storage of your tissue, we recommend storing frozen tissue blocks, or cryosections, at -20°C or -80°C.
For whole-mount immunostaining of zebrafish retinae, fixed larvae should be washed in PBS prior to starting. The fresher the tissue the better, but if you would prefer not to proceed with the staining protocol immediately, fixed larvae can be stored in PBS + sodium azide at 4°C for up to two weeks. The azide will prevent bacterial and fungal growth, which can be detrimental to the quality of your staining down the line.
Expert tip: If you find that your larvae are sticking to the pipette when washing, try using PBS with 0.1% Triton-X or Tween-20 (common detergents) instead. This will also help with tissue permeabilization and give you a head start in the lengthy series of washes.
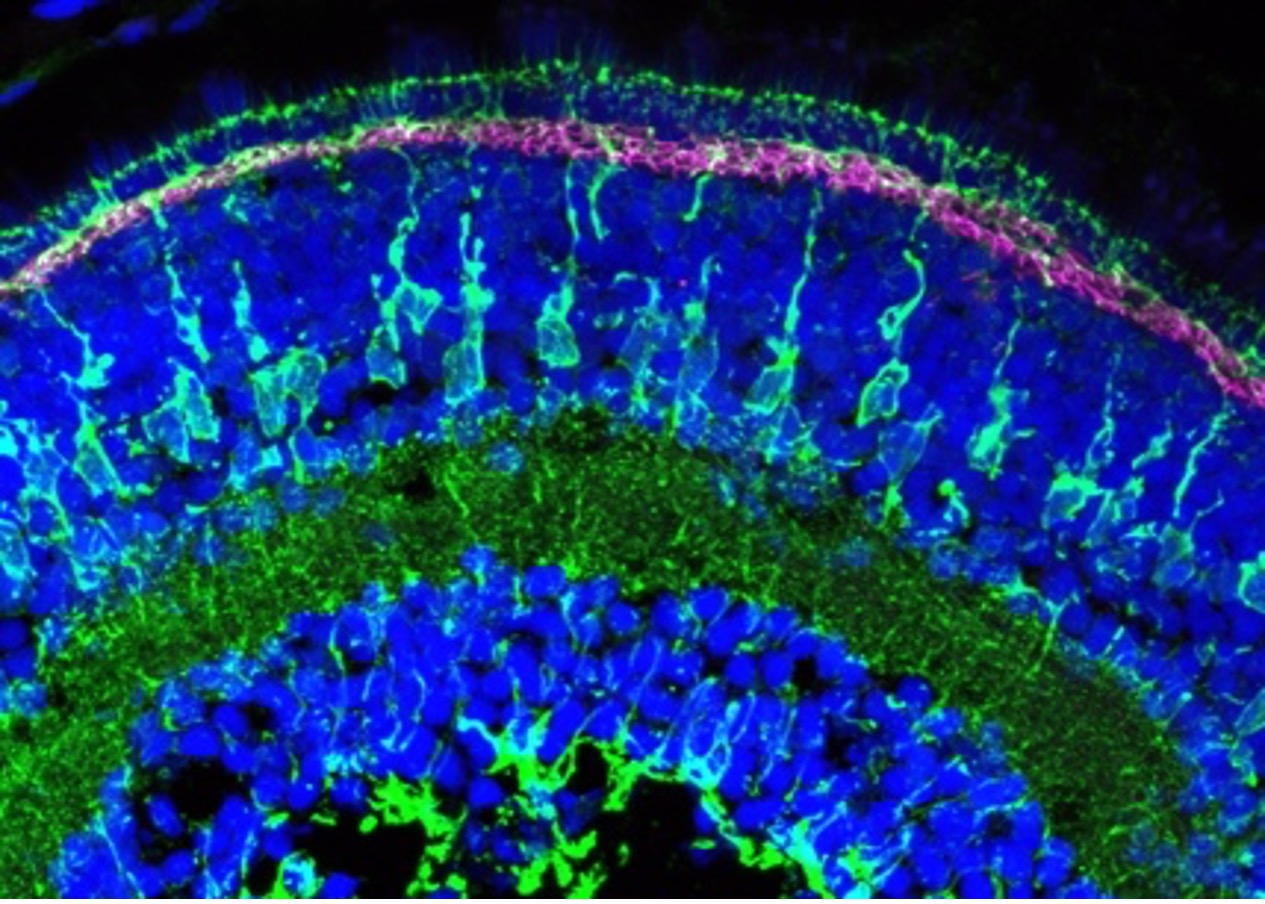
Figure 3: Immunofluorescence analysis of zebrafish retina at 5 days post fertilization. Collagen18a1 (aka Endostatin) polyclonal antibody (18301-1-AP, magenta). Dilution 1:200. Glutamine synthetase (66323-1-Ig, green) dilution 1:500. Tissue fixed overnight in 4% PFA; DAPI, blue. Data generated by Natalia Jaroszynska in Dr. Ryan MacDonald’s lab, University College London, UK.
Immunostaining
Step 1: Tissue permeabilization
This step involves the use of detergents such as Triton-X or Tween-20 to gently permeabilize the tissue or cells of interest to maximize the penetration of antibodies into the tissue. This is particularly important for the detection of intracellular antigens, as it permeabilizes cell membranes to allow antibodies to access intracellular proteins. For whole-mount IF, the wash incubations are performed in Eppendorf tubes and need to be much longer in duration since an intact retina is much denser than a tissue section.
Expert tip: Try increasing the concentration of your detergent from the standard 0.1% to 1% for whole-mount staining to improve the penetration of your antibodies. This is especially critical when working with thick, densely packed tissues like the retina.
Step 2: Antigen retrieval
Antigen retrieval is an important step in the IF protocol that is often omitted by labs. As the name suggests, this step maximizes the availability of your antigen for specific antibodies to bind. This can be achieved by heating your tissue sections, or whole-mount larvae, in an acidic (sodium citrate, pH 6) or basic buffer (Tris-HCl, pH 9), for 15–20 minutes prior to blocking and antibody incubations. Of course, each protein has a unique folding pattern, so how its 3D conformation will be affected by different pH conditions will vary. Hence, this step is entirely dependent on the specific protein you are detecting.
As a rule of thumb, perform a control experiment for every new antibody you use to compare the quality of your staining in tissues incubated with sodium citrate (pH 6), Tris-HCl (pH 9), versus no antigen retrieval. Once you know which antigen retrieval method is optimal for your antibodies of interest, start your antigen retrieval on your precious samples. For whole-mount IF, Willbrodt et al. have also shown that a 20-minute treatment with ice-cold acetone at -20°C can drastically improve the quality of your staining. Incubate the zebrafish in 100% ice-cold acetone in the freezer and wash off extensively with PBST after 20 mins.
Expert tip: Don’t have an incubator or oven to perform antigen retrieval on slides in? You can use a microwave, or like in our lab, use a simple rice cooker to achieve the best results – we stand by it! For whole-mount IF, place your larvae in antigen retrieval buffer in Eppendorf tubes and simply incubate them on a heat block at 70°C for 15 minutes, and don’t forget the extra acetone step straight after antigen retrieval!
Step 3: Antibody staining
Prior to antibody application, incubate slides in blocking solution for 1 hour at room temperature and whole larvae for 2 hours on a shaker. Then, use the same solution to prepare your primary antibody buffer containing your desired antibodies in blocking solution (10% goat serum, 1% BSA, 0.1% Triton-X in 1X PBS) and apply a small amount of antibody solution on the slide, enough to cover your sample. We recommend gently placing a rectangle of parafilm atop the antibody solution to help it spread homogenously, in addition to preventing evaporation overnight. To address the latter, we also always perform this step in a sealed, humidified chamber, which you can create by placing a damp paper towel or pouring some water at the bottom of a staining tray or a plastic slide box if you don’t have one. For whole-mount IF, we recommend incubating the zebrafish larvae in an Eppendorf tube at 4°C for at least 48 hours, particularly when staining thicker tissues like the retina. The following day, wash the antibodies off thoroughly before staining with the appropriate secondary antibodies, by incubating in PBST 3 times for 20 mins. After the secondary antibody incubation step, you must ensure that you protect your samples from the light using aluminum foil to prevent quenching of the fluorophores. After 2 hours, wash off your secondary antibodies thoroughly with 0.1% PBST, cover-slip your slides with your chosen mounting media, and you’re done!
Expert tip: It is important to be gentle when applying solutions (e.g., wash buffer) to the slides, as zebrafish larval sections are very delicate and small and can easily fall off, so don’t pipette anything directly onto the sections, but rather the spaces between them.
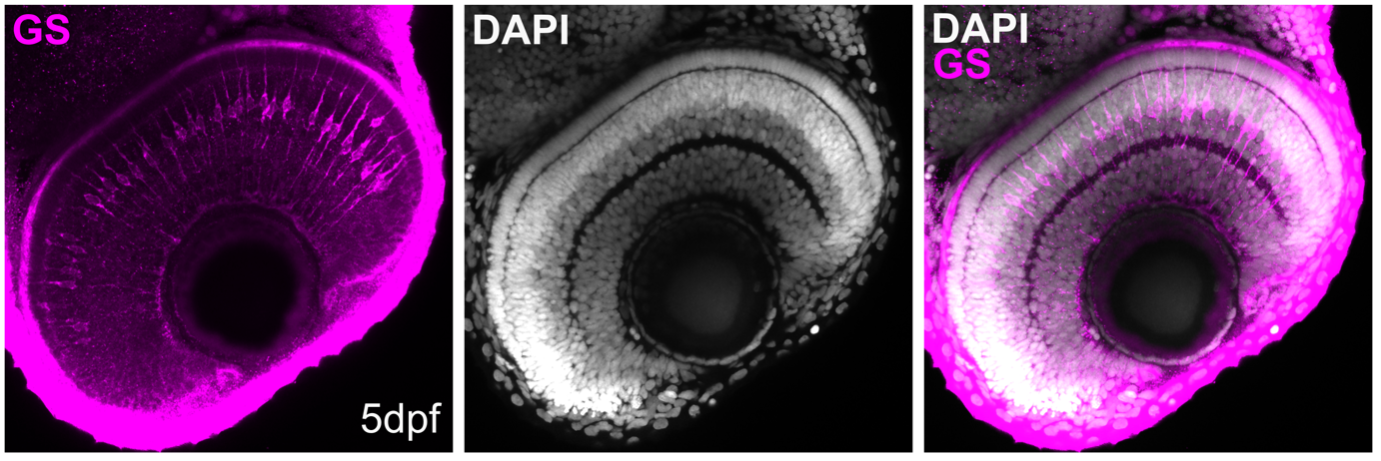
Figure 4: Whole-mount immunostaining for glutamine synthetase (GS; magenta; # 66323-1-Ig), on tissue sections of zebrafish retina at 5 days post fertilization (dpf). DAPI, white. Images acquired using a 40x objective lens, at 0.6X zoom. Dilution 1:500; Tissue fixed overnight in 4% PFA; Data generated by Natalia Jaroszynska in Dr. Ryan MacDonald’s lab, University College London, UK.
Imaging
Now onto the fun part, where we finally get to see the results! This step will depend on the type of fluorescent microscope available to you. Due to our thicker tissue samples, we use a confocal microscope that enables Z-stacking, but other types, such as a light sheet microscope, are excellent alternatives. Microscope selection will largely depend upon your requirements, the nature of your sample, and the resources available. Each microscope offers unique advantages, but a standard confocal microscope is our preferred option for imaging tissue sections as well as whole-mount zebrafish larvae at high resolution. For imaging cryosections on slides, we use the 40x objective to capture the details of the retina and use the optical zoom function to capture the full larval eye, or its specific regions. The 20x objective would allow you to image the whole larval head, but at the expense of resolution, so always tailor your imaging to the downstream applications.
For imaging whole-mount retinas using an inverted confocal microscope system, we first need to embed our stained larvae in a fixed and optimal position for imaging your tissue of interest. We recommend embedding your larvae in 1% low melting point agarose (w/v) in PBS on a specialized, commercially available imaging dish or multi-chamber slide, ensuring they have a glass bottom that is optimal for confocal imaging. To embed larvae for imaging, melt the agarose by heating aliquots on a heat block, and cooling down to ~40°C. You don’t want the agarose to cook your sample, but equally, you need it to be fluid enough to allow you time to orient the larvae before it sets. Transfer the larvae into the tube with agarose and gently mix. Use a Pasteur pipette to pick up the embryos and transfer them onto the glass part of the imaging dish, creating a dome of agarose. Next, use a small and precise tool, such as a microneedle loading tip, syringe needle, or fine forceps, to move the larvae into position. Remember to work quickly here as the agarose will set fast. Once set, pour PBS into the dish to prevent the agarose from drying out and let the embedded sample acclimate to the temperature of the microscope room or chamber before imaging, as this will prevent sample drifting during z-stack acquisition.
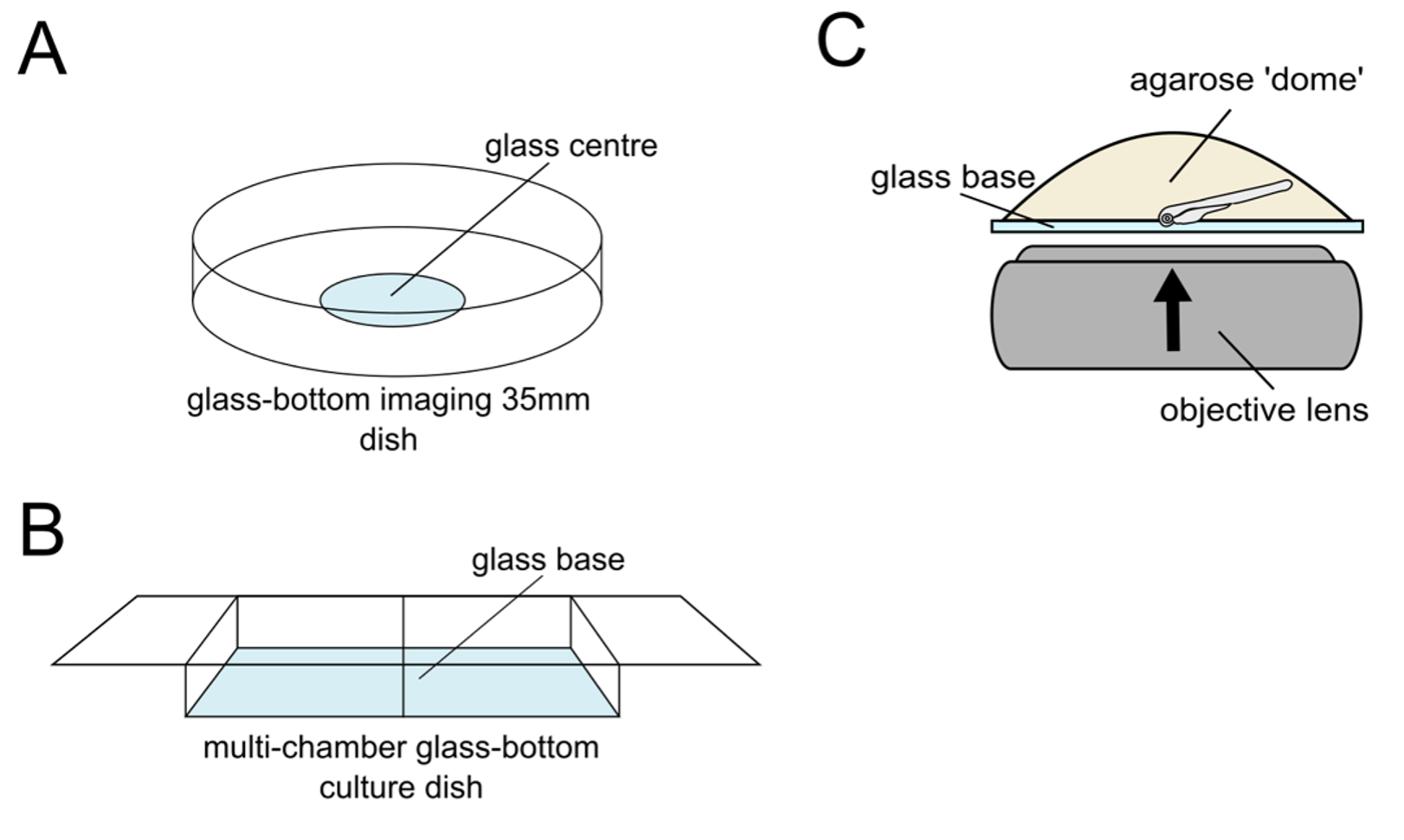 Figure 5: Mounting and imaging of larvae
Figure 5: Mounting and imaging of larvae
The acquisition parameters are a key component of your experiment. Detailed 3D analysis of your images will require higher resolution imaging through slower scan speeds and pixel averaging, as well as much smaller intervals between each slice in the z-stack. Tailor this part to your downstream applications and analyses. The most important aspect to remember is to fine-tune the laser power. Watch out for oversaturated pixels – you can always turn up the intensity of your image post-acquisition, but you cannot remove the signal.
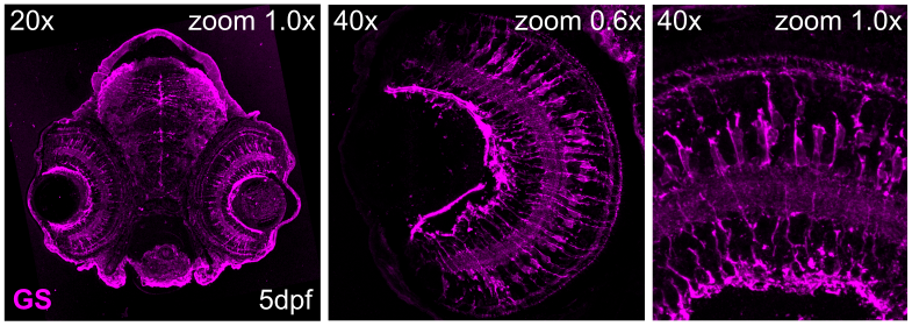
Figure 6: Immunostaining for glutamine synthetase (GS; # 66323-1-Ig), on tissue sections of zebrafish retina at 5 days post fertilization (dpf). Images acquired using 20x and 40x objective lens, at 1.0X or 0.6X zoom. Dilution 1:500; Tissue fixed overnight in 4% PFA; Data generated by Natalia Jaroszynska in Dr. Ryan MacDonald’s lab, University College London, UK.
Reference:
Inoue D, Wittbrodt J. One for all--a highly efficient and versatile method for fluorescent immunostaining in fish embryos. PLoS One. 2011;6(5):e19713. doi: 10.1371/journal.pone.0019713. Epub 2011 May 13. PMID: 21603650; PMCID: PMC3094454.

Related Content
How to choose the right secondary antibody?
A Guide to Staining Organelles
Want to upgrade your immunofluorescence workflow? Go Direct!
Imaging the Rainbow: Multiplexing with Same-Species Antibodies for Immunofluorescence
Support
Newsletter Signup
Stay up-to-date with our latest news and events. New to Proteintech? Get 10% off your first order when you sign up.